13.1 Exercise 2 - Barplots
- Import GSE277039/gencode.vM38.annotation_sample.csv in an object that you will call gtf. You can check the first 20 rows of gtf using the head() function: check the help page to see how it works.
correction
The data in gtf represents a small subset of the gencode vM38 mouse gene annotation.
## # A tibble: 20 × 7
## chr start end strand gene_id gene_type gene_symbol
## <chr> <dbl> <dbl> <chr> <chr> <chr> <chr>
## 1 chr10 41361638 41366365 - ENSMUSG00000019822.13 protein_coding Smpd2
## 2 chr19 3901773 3944369 + ENSMUSG00000024843.16 protein_coding Chka
## 3 chr4 37297905 37298025 + ENSMUSG00000088718.4 snoRNA Gm22732
## 4 chr9 99125420 99182457 + ENSMUSG00000056267.15 protein_coding Cep70
## 5 chr14 18063426 18072247 - ENSMUSG00000095056.8 protein_coding Gm3159
## 6 chr7 135886020 135901318 - ENSMUSG00000127332.1 lncRNA Gm72246
## 7 chr16 34470291 34498988 + ENSMUSG00000022832.12 protein_coding Ropn1
## 8 chr11 46345762 46372082 + ENSMUSG00000020399.15 protein_coding Havcr2
## 9 chr6 5767578 5768992 - ENSMUSG00000136477.1 lncRNA Gm67521
## 10 chr7 42258950 42292012 - ENSMUSG00000074158.10 protein_coding Zfp976
## 11 chr15 102426627 102432111 + ENSMUSG00000023051.13 protein_coding Tarbp2
## 12 chr12 85253391 85255602 - ENSMUSG00000126100.1 lncRNA Gm59518
## 13 chr9 109946776 110069246 + ENSMUSG00000032481.18 protein_coding Smarcc1
## 14 chr3 94882042 94914154 + ENSMUSG00000038861.15 protein_coding Pi4kb
## 15 chr15 83960298 83989955 - ENSMUSG00000018865.10 protein_coding Sult4a1
## 16 chr13 48298781 48310585 + ENSMUSG00000127329.1 lncRNA Gm52065
## 17 chr17 46875397 46940430 - ENSMUSG00000023972.11 protein_coding Ptk7
## 18 chr5 143950816 143950919 + ENSMUSG00000119602.1 snRNA Gm24311
## 19 chr7 41485238 41522136 - ENSMUSG00000090383.3 protein_coding Vmn2r58
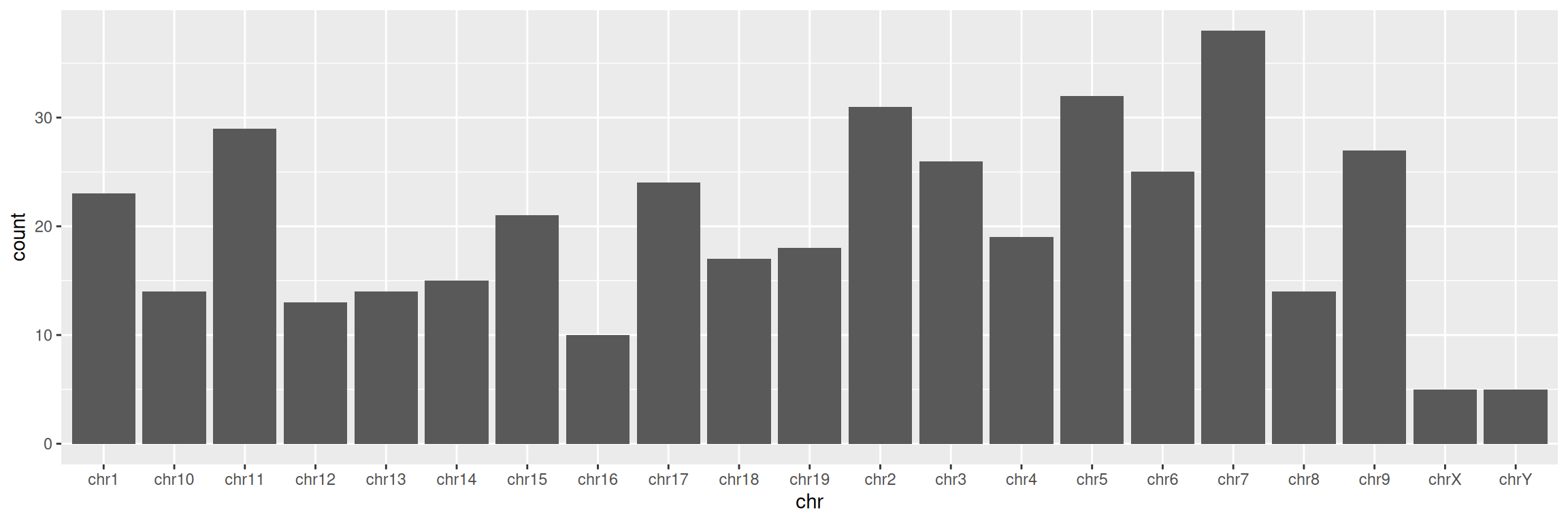
## 20 chr3 89344013 89358259 + ENSMUSG00000042613.10 protein_coding Pbxip1- Create a simple barplot displaying the number of genes per chromosome (tip: you want to visualize each chromosome on the x axis):
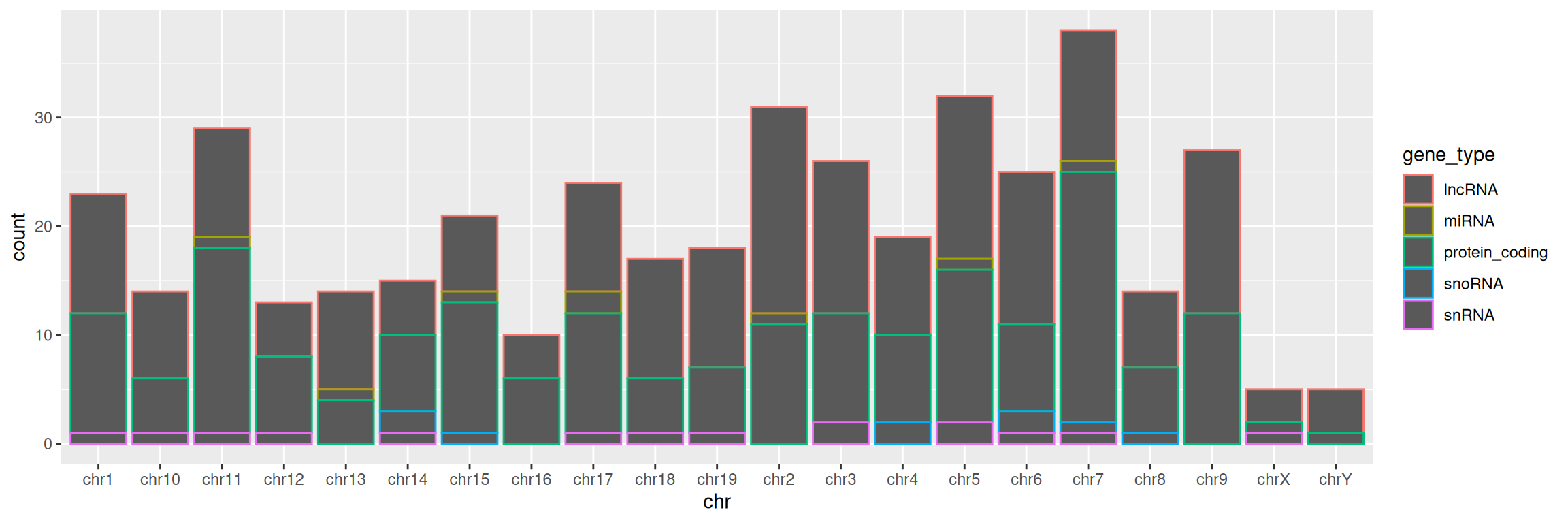
3. Keep chromosomes on the x axis, and split the barplot per gene type.
TIP: remember how we set color= in mapping=aes() function in the scatter plot section? Give it a try here!
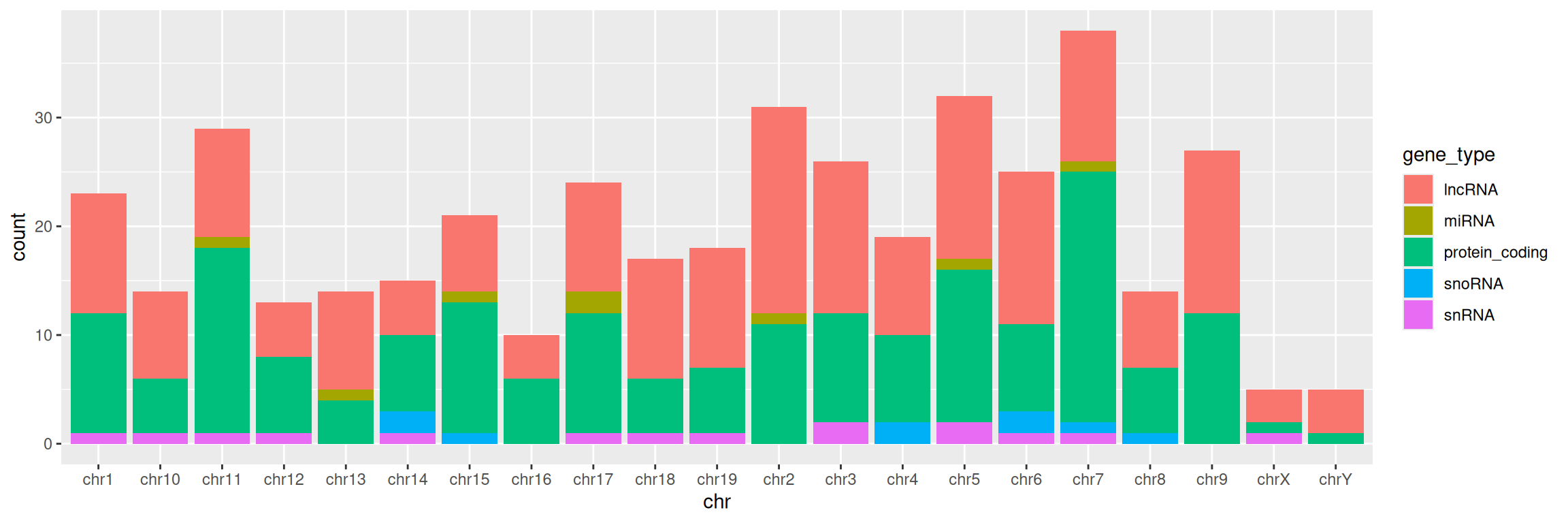
4. Change color= with fill= in aes(). What changes?
5. Add a title to the graph:
correction
ggplot(data=gtf, mapping=aes(x=chr, fill=gene_type)) +
geom_bar() +
ggtitle(label = "Number of genes per chromosome, split by gene type")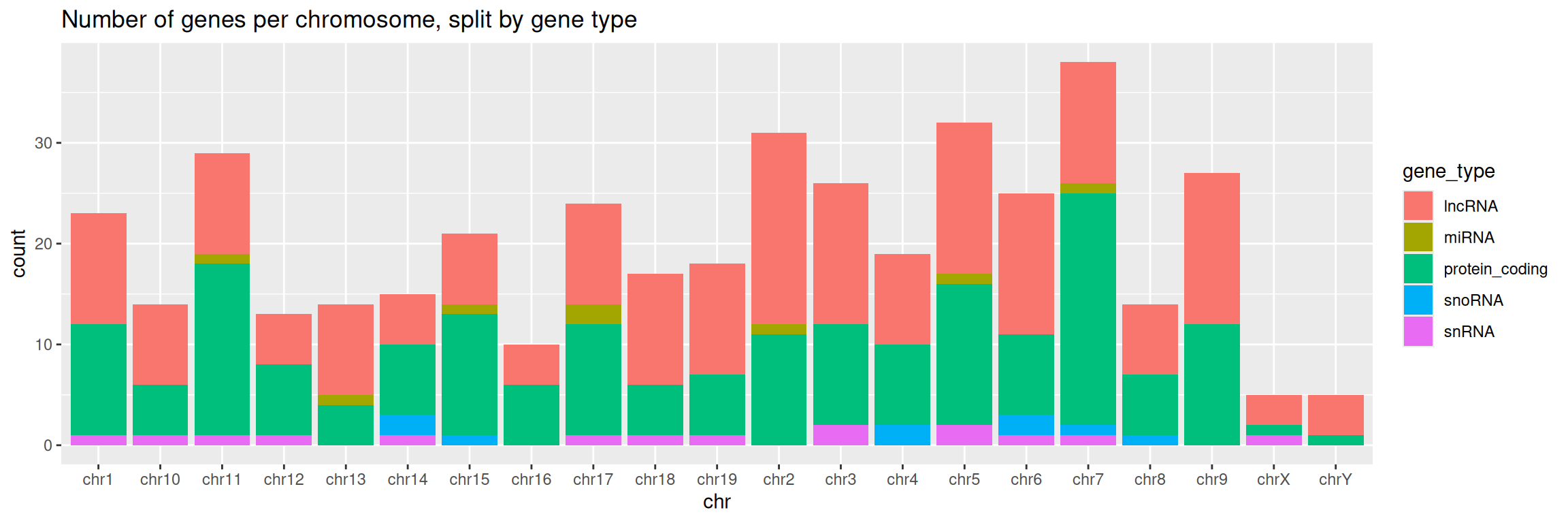
6. Change the default theme:
correction
ggplot(data=gtf, mapping=aes(x=chr, fill=gene_type)) +
geom_bar() +
ggtitle(label = "Number of genes per chromosome, split by gene type") +
theme_bw()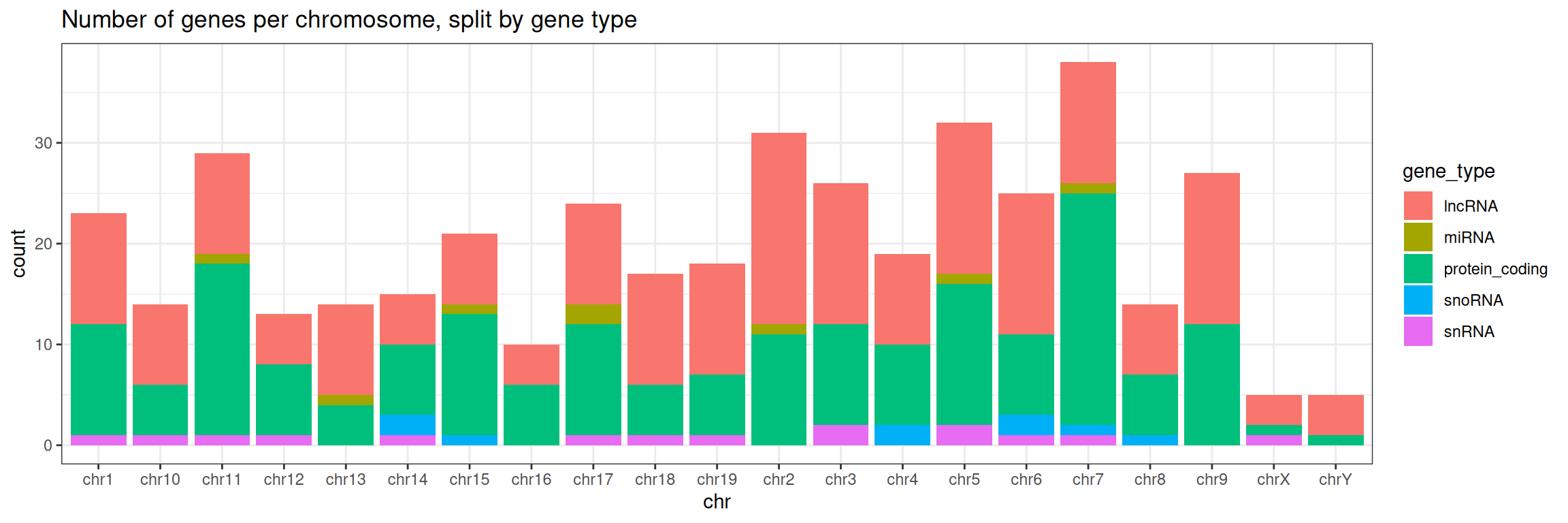
7. Save the graph in PNG format in the workshop’s directory.